Overview: N-terminus and C-terminus in Protein
Proteins are composed of linear chains of amino acids, with each protein having two distinct ends: the N-terminus, featuring a free amine group, and the C-terminus, having a free carboxyl group. These termini are essential for the protein's function, stability, and interaction with other molecules. The analysis of these termini is crucial in protein engineering, drug development, and quality control of biopharmaceuticals.
Significance of N-terminal and C-terminal Analysis
Research on Protein Physiological Functions
Analyzing the N-terminus and C-terminus of proteins is crucial due to the significant roles these terminal regions play in protein functionality, localization, and stability. The diversity of protein forms generated from a single gene through mechanisms such as co- or post-translational modifications, alternative splicing, and alternative translation initiation makes terminal analysis indispensable for understanding protein biology.
Verification of Protein Terminal Modifications
Precise chemical modification of proteins can improve their physicochemical properties and endow them with new physiological functions, such as extending protein half-life and regulating protein interactions. The N-terminus of a protein or peptide drug is crucial as it determines the site and efficacy of the drug's action. Changes at the N-terminus, including alterations and modifications of the N-terminal amino acids during the drug manufacturing process or storage, are common. In contrast to the development of modification techniques for protein side chains and N-termini, precise strategies for C-terminal modifications are still significantly lacking. During the modification process of protein N-termini or C-termini, it is essential to monitor and verify their sequences to obtain correctly modified samples.
Terminomics Analysis (High Throughput)
With the rise of proteogenomics, re-annotating genomes using proteomics data has become a research hotspot. Employing proteomics methods for large-scale identification and analysis of protein termini helps verify and correct annotated genes and even discover new genes. Over the past 20 years, mass spectrometry technology has undergone rapid development, leading to more in-depth studies on the proteomics of protein termini. The establishment and improvement of various terminal enrichment techniques and methods have gradually matured large-scale sequencing of protein termini.
Quality control of biopharmaceutical products
Analyzing the N- and C-termini is vital across a broad spectrum of biological products, including proteins, antibodies, vaccines, polypeptides, and recombinant collagen. Such analysis ensures the structural integrity and biological activity of these products. For instance, in monoclonal antibody (mAb) quality control, identifying and quantifying terminal amino acid sequences in light and heavy chains is crucial. Multiple terminal sequences must be accurately determined to ensure product consistency and efficacy.
Methods of N-terminal and C-terminal Analysis
Advances in proteomics have introduced diverse technologies for terminal analysis, each offering unique benefits and insights.
Currently, there are two primary methods for N-terminal sequencing of proteins.
The first method is Edman degradation, which sequentially cleaves and analyzes the limited number of amino acids from the N-terminus. This method determines the N-terminal amino acid sequence by comparing the retention times of amino acid standards.
The second method is mass spectrometry, where the protein is denatured, reduced, and enzymatically digested into peptides of varying sizes. These peptides are then separated by liquid chromatography and introduced into a high-resolution mass spectrometer at different time intervals. The m/z values of the peptides are analyzed and compared against a proposed sequence.
Depending on the conditions and the amount of amino acid data required, these two methods are often combined or chosen based on comprehensive considerations.
Principle
Edman degradation is a process for determining the amino acid sequence of a peptide starting from the free N-terminus. Under alkaline conditions, phenylisothiocyanate (PITC) reacts with the N-terminal amino group of the protein or peptide to form a phenylthiocarbamoyl (PTC) derivative. This derivative is then treated with acid, causing cyclization and selective cleavage of the N-terminal residue, producing a phenylthiohydantoin (PTH) derivative of the N-terminal amino acid. The cyclic peptide then enters the next cycle. The PTH derivative is extracted with an organic solvent, and under acidic conditions, it reacts to form a stable phenylthiohydantoin (PTH) derivative. This PTH derivative is analyzed by HPLC to identify the amino acid. Each cycle provides the information of one amino acid.
Procedure
- Sample Preparation: Before protein sequencing, the protein sample is separated by SDS-PAGE to ensure purity. The protein on the SDS-PAGE is then transferred to a PVDF membrane. After staining, the protein bands are cut out and analyzed using a protein sequencer.
- Analysis: Edman degradation sequencing involves cyclic reactions that sequentially identify amino acids from the N-terminus of the protein. Each Edman sequencing reaction consists of three steps: first, PITC reacts with the free amino group at the N-terminus under alkaline conditions; second, the N-terminal residue is cleaved in an acidic solution; third, the PTC derivative is converted into a more stable PTH derivative, which is then analyzed by HPLC to determine the type of amino acid based on its retention time.
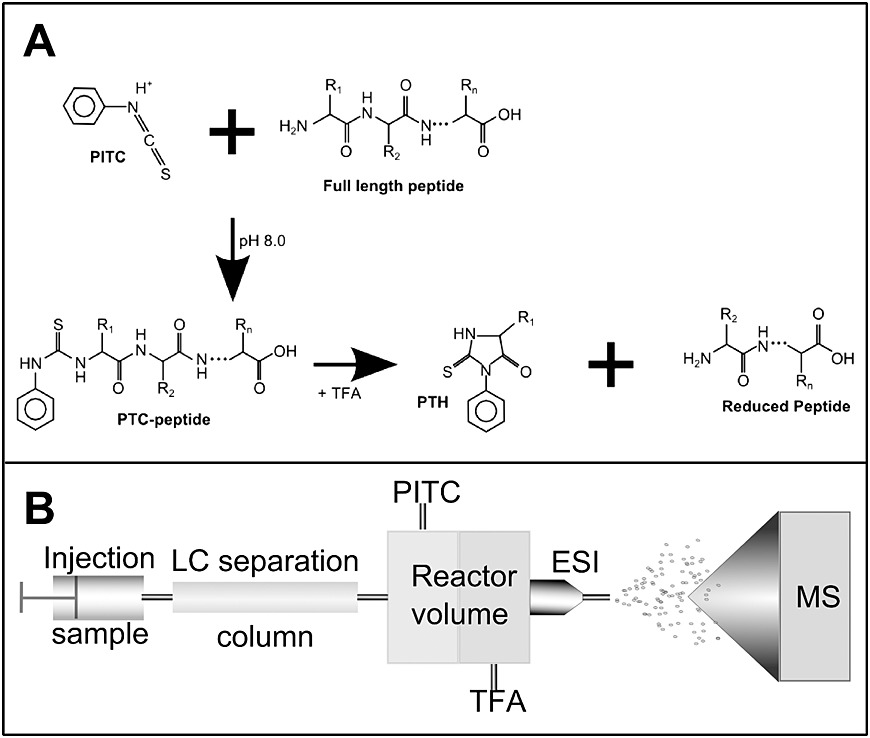 Figure 1. Combination of Edman degradation with LC-MS workflow. (Anna A. et al., 2013)
Figure 1. Combination of Edman degradation with LC-MS workflow. (Anna A. et al., 2013)
Instrumentation
Based on the steps of Edman amino acid sequencing, instrument companies have developed protein sequencers, which include two main modules: a sample processing module and a liquid chromatography module. The sample processing module automates coupling, cleavage, extraction, and conversion, while the liquid chromatography module automatically analyzes the obtained PTH-amino acids, identifying the amino acid types.
Advantages
- High Reaction Yield and Recovery: PITC reacts efficiently with all amino acid residues, resulting in high yield and recovery rates. Consequently, the formation of by-products is minimal, allowing for accurate identification through chromatography.
- Fast Reaction Time: The Edman degradation process is relatively quick. For most amino acid residues, the coupling reaction requires only about 30 minutes, and the cleavage reaction can be completed in just 5 minutes.
- Retention of Intact Peptide Chains: After the reaction, the peptide chain remains intact, enabling repeated Edman degradation cycles to determine newly exposed N-terminal amino acids. This allows for sequential and thorough analysis of the N-terminal sequence.
Disadvantages
- Limitations with Modified or Blocked N-Termini: Peptides that have undergone chemical modifications or have blocked N-termini cannot be effectively analyzed using Edman degradation.
- Low Throughput: Edman degradation is classified as a low-throughput method. While it is reliable for sequencing, it can only analyze approximately 30 amino acids in a single run when sample quantities are adequate.
Mass Spectrometry (MS)
Principle
The most widely used method for protein sequencing currently is mass spectrometry (MS). MS-based protein sequencing strategies are broadly categorized into two main approaches: Top-Down and Bottom-Up.
Top-Down Strategy: This approach analyzes intact proteins directly using liquid chromatography-mass spectrometry (LC-MS) without prior degradation. The protein's sequence is determined by identifying fragment ions in the mass spectra.
Bottom-Up Strategy: In this approach, proteins are first hydrolyzed into peptides. These peptides are then analyzed using LC-MS, where they are sequenced de novo and their sequences are pieced together to reconstruct the complete protein sequence.
Procedure
Top-Down Approach
- Separation and Ionization: Intact proteins are separated from complex biological samples using reverse-phase liquid chromatography. The proteins are ionized using electrospray ionization (ESI) or matrix-assisted laser desorption/ionization (MALDI).
- Fragmentation: The resulting ions are fragmented through collision-induced dissociation (CID), high-energy collision dissociation (HCD), electron capture dissociation (ECD), or electron transfer dissociation (ETD).
- MS Analysis: The fragments are analyzed by tandem mass spectrometry (MS/MS).
Bottom-Up Approach
- Digestion: Protein mixtures are digested into peptide mixtures. The peptide mixtures are separated by chromatography and ionized.
- MS Analysis: The ionized peptides are analyzed by tandem mass spectrometry (MS/MS) to generate peptide fingerprints, which are used for peptide identification.
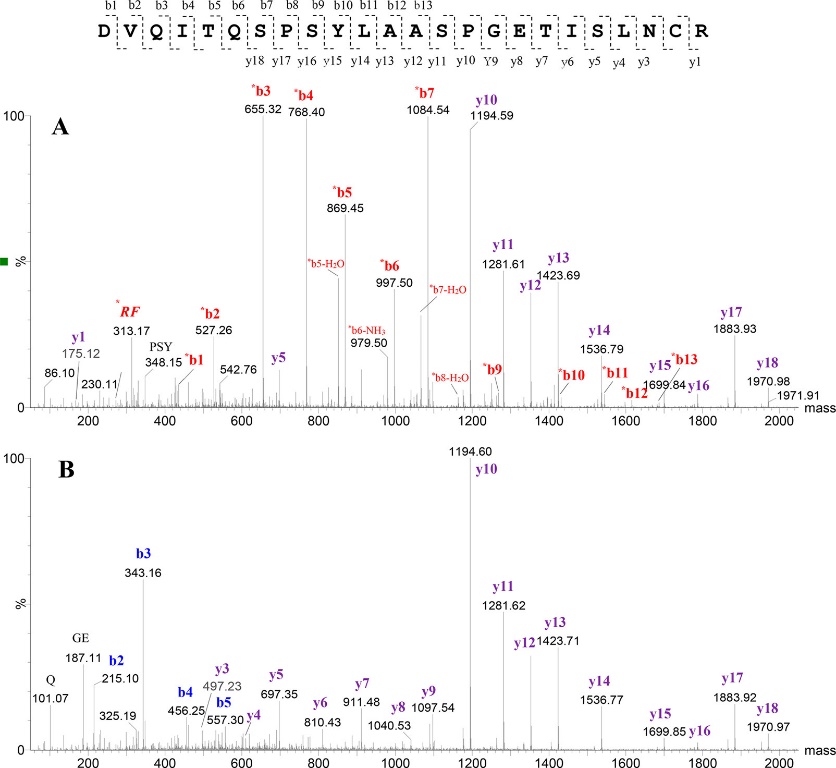
Figure 2. CID MS/MS spectra of the N-terminal tryptic peptide of the light chain for mAb1. (Malgorzata Monika et al., 2019)
Instrumentation
High-performance liquid chromatography-tandem mass spectrometry (LC-MS) consists of two main components:
- High-Performance Liquid Chromatography (HPLC): Used for the separation of proteins.
- Tandem Mass Spectrometry (MS/MS): Used for ionizing proteins and analyzing their fragments.
Advantages
- N-Terminal Sequencing with High-Resolution Mass Spectrometry: This method allows for the analysis of N-terminal blocking and post-translational modifications (PTMs) through high-resolution mass spectrometry, determining the protein's N-terminal starting point.
- Complex Protein Analysis: By analyzing the labeling positions (such as N-terminal dimethylation labeling), it is possible to determine the N-terminal starting points of multiple proteins in a complex protein sample.
- Identification of Non-Standard Amino Acids: Mass spectrometry can identify non-standard amino acids that are difficult to determine using traditional chemical methods due to the lack of reference standards.
Disadvantages
- Database Dependence: Mass spectrometry identification often relies on database comparison for protein sequence analysis. Therefore, proteins not included in the database cannot be accurately analyzed by mass spectrometry.
- Incomplete Sequence Coverage: Under current conditions, mass spectrometry rarely provides 100% sequence coverage of peptides. Some peptide fragments may be missed.
- Interference Issues: The accuracy of analysis can be affected by protein contamination, isotopic peak interference, and incomplete peptide fragmentation.
Carboxypeptidase Digestion
Principle
Carboxypeptidases are exopeptidases that specifically hydrolyze peptide bonds at the C-terminus of proteins, releasing amino acids one at a time. The number and type of amino acids released vary with reaction time. By analyzing the amount of amino acids released over time, the sequence of amino acids at the C-terminus can be determined.
Procedure
- Enzymatic Digestion: The enzyme cleaves amino acids sequentially from the C-terminus, releasing free amino acids over time.
- Analysis: The identity of the peptide sequence can subsequently be deduced by HPLC-based detection of released residues (considering the release order) or by analyzing the truncated peptides using MALDI-TOF-MS.
Instrumentation
The analysis method depends on the approach used:
- High-Performance Liquid Chromatography (HPLC): Used for direct detection of amino acids released by hydrolysis.
- Mass Spectrometry (MS): Analyze the difference in molecular weight between the original protein or peptide and the truncated fragments to determine the molecular weight of the released amino acids.
Advantages
- High Molecular Weight Protein Analysis: By combining the chemical cleavage of proteins with cyanogen bromide and the specific degradation of the C-terminus using carboxypeptidases, it is possible to use mass spectrometry to detect the peptide fragments obtained at different digestion times.
- Enhanced Precision and Sensitivity: The catalytic hydrolysis conditions of carboxypeptidase can be optimized by collecting data at different digestion time points, thereby improving the accuracy and sensitivity of the analysis.
Disadvantages
- High Cost of Optimization: Since the cleavage rate of carboxypeptidase largely depends on the peptide sequence, this method usually requires careful optimization of reaction conditions, making it labor-intensive and requiring relatively large amounts of sample.
Mass Spectrometry (MS)
Principle
MS-based methods for C-terminal sequencing of proteins utilize two main approaches: top-down and bottom-up. The bottom-up approach is widely used due to its practical advantages, including higher sensitivity for low-abundance proteins, easier handling of complex protein mixtures, and compatibility with existing proteomic workflows. In the bottom-up approach, proteins are enzymatically digested into peptides, which are then analyzed by mass spectrometry (MS). It can further be categorized into two main strategies:
Labeling Strategies
Labeling strategies involve chemically modifying peptides to introduce tags or labels that aid in their detection and sequencing by MS. These strategies enhance the ionization efficiency and distinguish C-terminal peptides from other peptide fragments in the mixture. Examples include:
- 18O-Labeling: Incorporation of 18O atoms at the C-terminal carboxyl group during protease digestion, allowing for differential mass tagging.
- Isotopic Labeling: Introduction of stable isotopes (e.g., 13C or 15N) into peptides, enabling quantitative analysis and identification of C-terminal peptides.
Enrichment strategies
Enrichment strategies focus on isolating C-terminal peptides from complex peptide mixtures before MS analysis. This selective enrichment improves sensitivity and specificity in identifying C-terminal sequences. Common enrichment methods include:
- Positive Selection: Direct targeting and isolation of C-terminal peptides using affinity tags or antibodies specific to C-terminal sequences.
- Negative Selection: Depletion of non-C-terminal peptides, leaving enriched C-terminal peptides for subsequent MS analysis.
Procedure
The methods encompassed within Labeling Strategies and Enrichment Strategies are highly diverse. Here, two straightforward approaches are outlined respectively:
Labeling Strategies
- Digestion: Enzymatic digestion of proteins is conducted in a buffer solution containing H218O, leading to the incorporation of one or two oxygen-18 atoms by the protease at the carboxyl moiety of the cleaved peptide bond. This process does not modify the carboxyl group of the original C-terminal peptide.
- MS Analysis: In MS1 analysis, C-terminal peptides appear as singular peaks, whereas N-terminal and internal peptides exhibit an isotope envelope reflecting the incorporation of 18O atoms (resulting in a 2 or 4 Da mass shift).
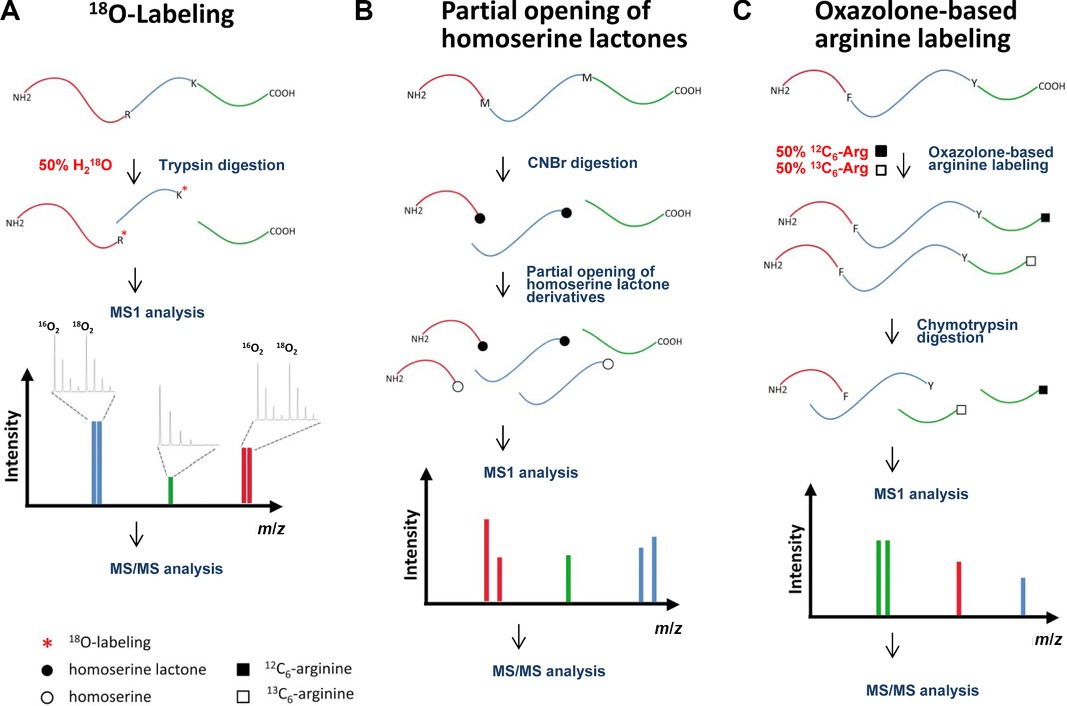
Figure 3. Schematic representation of methods for C-terminal labeling. (A) Protease-assisted 18O-labeling. (B) Method to differentiate C-terminal peptides in cyanogen bromide digests. (C) Isotopic arginine labeling based on the oxazolone chemistry. (Sebastian et al., 2015)
Enrichment strategies
The COFRADIC technology exploits peptide chromatography to specifically isolate peptides of interest from complex peptide mixtures.
- Digestion: After acylating all primary amines, the proteome is subjected to trypsin digestion.
- Separation: Strong cation exchange (SCX) fractionation is carried out under acidic conditions (pH 3.0) on the tryptic digest. An additional step involves chemical derivatization of primary amines present in C-terminal peptides using an N-hydroxysuccinimide ester of butyrate. This step is performed between two consecutive RP-HPLC separations to further segregate C-terminal peptides from N-terminal peptides.
- MS Analysis: The enriched C-terminal peptides are analyzed using LC-MS/MS instrumentation.
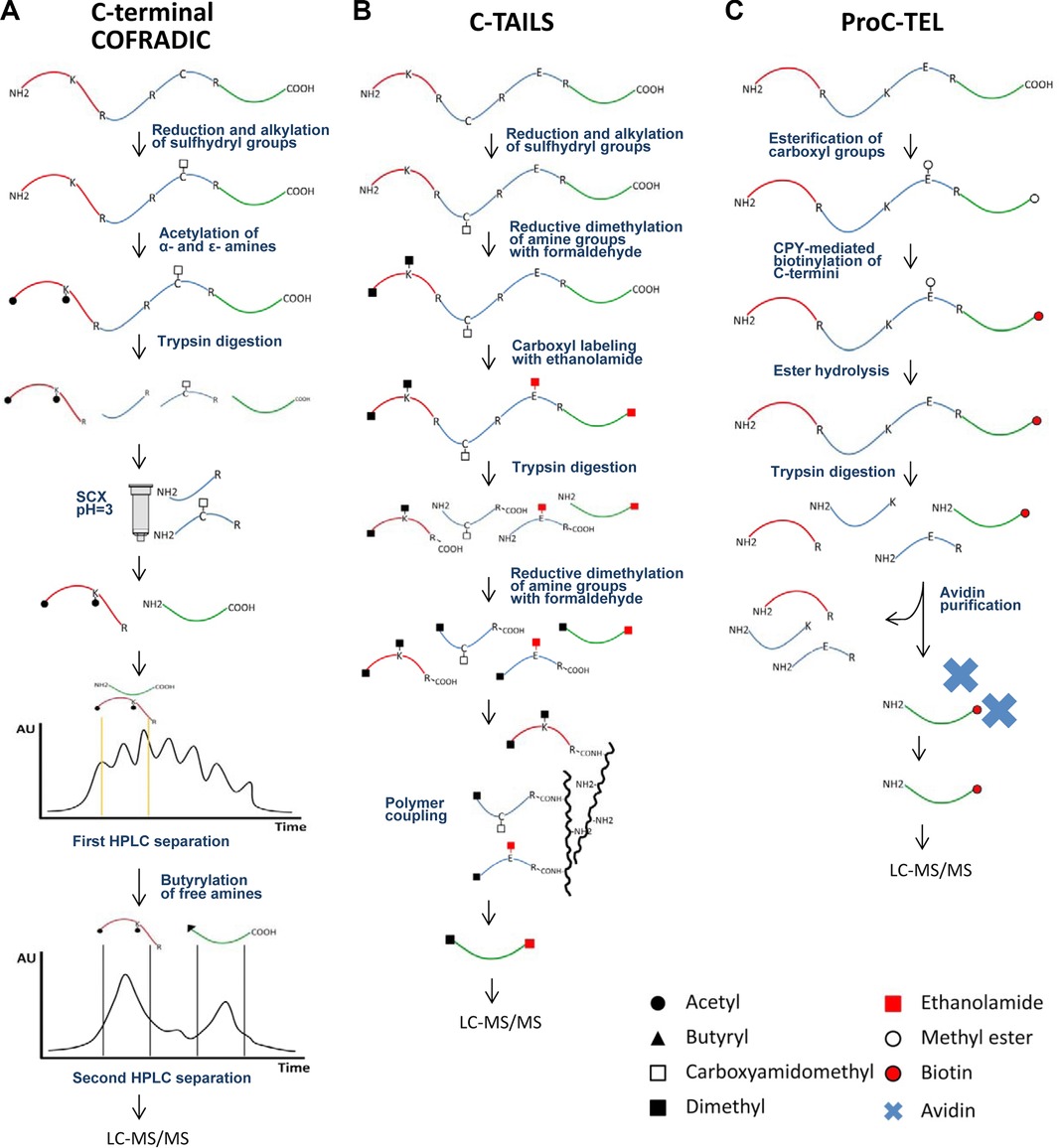
Figure 4. Schematic representation of C-terminal enrichment strategies. C-terminal COFRADIC (A) and C-TAILS (B) represent negative selection strategies for C-terminal peptide enrichment. On the other hand, ProC-TEL (C) applies a positive selection to isolate C-termini. (Sebastian et al., 2015)
Instrumentation
MS-based C-terminal sequencing requires advanced instrumentation:
- High-resolution Mass Spectrometers: Such as Time-of-Flight (TOF) instruments, for accurate mass measurement of peptides.
- Liquid Chromatography (LC): Coupled with MS (LC-MS), for peptide separation and enhanced resolution.
Advantages
- Enhanced Recognition Efficiency: C-terminal peptides typically represent only a small fraction of all peptides generated from protein digestion. Methods involving labeling or enrichment improve the efficiency of identifying C-terminal peptides.
- Improved Sensitivity and Accuracy: Mass spectrometry methods offer higher sensitivity and accuracy and are more cost-effective compared to enzymatic digestion approaches.
Disadvantages
- Incomplete Sequence Coverage: Under current conditions, mass spectrometry rarely provides 100% sequence coverage of peptides. Some peptide fragments may be missed.
Conclusion
Understanding the N-terminus and C-terminus is fundamental in the field of protein biochemistry. At Creative Proteomics, we leverage this knowledge to explore innovative solutions in biotechnology, enhancing protein function, stability, and interaction capabilities. The distinct roles and structural characteristics of these termini underscore their importance in protein biology, providing a foundation for advanced research and application in various scientific and industrial domains.
References
- Lobas, Anna A. "Combination of Edman Degradation of Peptides with Liquid Chromatography/Mass Spectrometry Workflow for Peptide Identification in Bottom-up Proteomics." (2013).
- Vecchi, Malgorzata Monika. "Identification and Sequencing of N-Terminal Peptides in Proteins by LC-Fluorescence-MS/MS: An Approach to Replacement of the Edman Degradation." (2019).
- Tanco, Sebastian. "C-Terminomics: Targeted Analysis of Natural and Posttranslationally Modified Protein and Peptide C-Termini." (2015).